
Cancer is a genetic disease characterized by uncontrolled cell growth and invasion that leads to uncontrolled proliferation and the potential for metastasis (Cooper, 2000). Genetic changes such as the mutation of BRCA1 and BRCA2 and the amplification of HER2 can result in the development of breast cancer (Jacobs et al., 2022). HER2 receptor overexpression (HER2+) due to the amplification of a segment of genes containing the HER2 gene in chromosome 17 occurs in around 20% of all breast cancers and is thus a known pharmaceutical target (Krishnamurti & Silverman, 2014). In 2020, 2.3 million new cases of breast cancer were diagnosed worldwide and approximately 685,000 patients died from breast cancer. Breast cancer accounts for about 25% of all female cancer cases and around 16% of all cancer deaths in the world (S. Lei et al., 2021). The beginning of breast cancer starts with stage 0 (Table 1) where the tumor is non-invasive. In stage 1, the tumor remains small and shows minimal spread to lymph nodes. In stage 2, the cancer cells have spread to nearby lymph nodes but not to other parts of the body. In stage 3, the tumor further increases in size and spreads to more lymph nodes. Stage 4 is the final stage where the cancer is metastatic and tumors have spread to distant organs such as the liver, lungs, and bones (Akram et al., 2017). HER2+ Breast cancer can be treated through surgery, radiation therapy, chemotherapy, and targeted therapy. Combining therapies with different mechanisms of action is often employed to maximize therapeutic benefit (Jacobs et al., 2022).
Causes of Cancer
Cancer is a genetic disease characterized by uncontrolled cell growth and genetic mutations play a crucial role in cancer development (Cooper, 2000). Cancer presents significant challenges in treatment and poses substantial threats to the body due to the ability of tumor cells to evade the immune response. Such evasion mechanisms include modifying surface antigens and manipulating the surrounding environment (Wang et al., 2017). Cancer is not the result of one mutation but rather caused by a complex and long series of genetic changes. These changes enable cells to acquire some cancerous traits and together cause the formation of cancer cells. Changes in two types of genes that regulate the cell cycle can lead to cancerous traits—proto-oncogenes and tumor suppressor genes. Proto-oncogenes encourage cell division and can mutate and become oncogenes that stimulate excessive cell division. Tumor suppressor genes inhibit cell division and its mutation allows aberrant cells to grow excessively. Collectively, mutations in these two categories of genes account for most causes of cancer (National Institutes of Health (US), 2007).
Tumor Suppressor Genes
Tumor suppressor genes play a crucial role in regulating various cellular functions such as cell growth, cell cycle progression, DNA repair mechanisms, and apoptosis induction. Loss of function mutations in these genes is a well-known mechanism that leads to the development of cancer. Tumor suppressor genes can be classified into five functional types depending on the type of protein it encodes. These five types of proteins are intracellular proteins, receptors, proteins involved in apoptosis induction, proteins involved in DNA repair, and checkpoint-control proteins. Intracellular proteins such as pRB and p16 are crucial in controlling the progression of cell cycle stages. Receptors such as adenomatous polyposis coli (APC) and transforming growth factor (TGF)-β orchestrate signals to inhibit cell proliferation. Proteins such as p53 are involved in the induction of apoptosis. Proteins that aid in the repair of DNA include DNA mismatch repair protein 2 (MSH2) and p53. Lastly, checkpoint-control proteins such as p16, p14, and breast cancer type 1 susceptibility protein (BRCA1) trigger cell cycle arrest in the event of DNA damage or chromosomal defects (Joyce et al., 2022). BRCA1 and BRCA2 are tumor suppressor genes that play critical roles in DNA repair, particularly in homologous recombination. Mutations in BRCA1 can impair homologous recombination, compromising DNA repair processes. BRCA2 regulates the activity of Rad51, an enzyme involved in homologous recombination. BRCA1 and BRCA2 are involved in several cellular processes, such as cell cycle control, gene regulation, and apoptosis (F. Lei et al., 2011). BRCA1 and BRCA2 mutations are hereditary and account for approximately 5% of all breast cancers (De Talhouet et al., 2020). Due to the higher occurrence and the greater variety of treatment options for HER2 overexpressed (HER2+) breast cancer, this article will focus on HER2+ breast cancer only.
Proto-oncogenes and Oncogenes
In addition to mutations of tumor suppressor genes, mutations of proto-oncogenes are also known genetic drivers of breast cancer. Proto-oncogenes are normal genes that play a crucial role in regulating cell growth and proliferation in normal cells. However, changes in their expression have the potential to contribute to cancer development. Proto-oncogenes can be converted into cancer-causing genes (oncogenes) through a series of events that alter their expression. These events include mutations of nucleic acid sequences, chromosomal rearrangements, and amplification of the number of genes. These alterations cause uncontrolled cell growth and proliferation that leads to cancer. Understanding the mechanisms that activate proto-oncogenes is critical to developing targeted therapies for different types of cancer. By identifying and targeting the specific genes responsible for cancer development, researchers can develop more effective and personalized treatments, leading to better outcomes for cancer patients (Cline, 1987).
HER2 in Breast Cancer
Human Epidermal Growth Factor Receptor-2 (HER2) is a proto-oncogene encoding receptor proteins that are involved in molecular signaling pathways and process growth-stimulating signals between cells (Ross et al., 2003). The amplification of HER2 is one of the primary causes of breast cancer and is found in around 20% of all breast cancers. Being a valuable therapeutic target, the 2007 American Society of Clinical Oncology guidelines require the evaluation of HER2 amplification in every invasive breast cancer (Krishnamurti & Silverman, 2014).
The HER2 gene is located in a segment of chromosome 17 and the amplification of this chromosome segment is almost always the cause of HER2 receptor overexpression in breast cancers. Breast cancers can have up to around 50 copies of the HER2 gene, which is an almost 100-fold increase, and around 2 million protein receptors expressed on the surface of a tumor cell. HER2+ breast cancers have distinguishable characteristics such as resistance to a few hormonal agents, increased sensitivity to some cytotoxic chemotherapy agents, and a larger brain metastasis probability (Moasser, 2007).
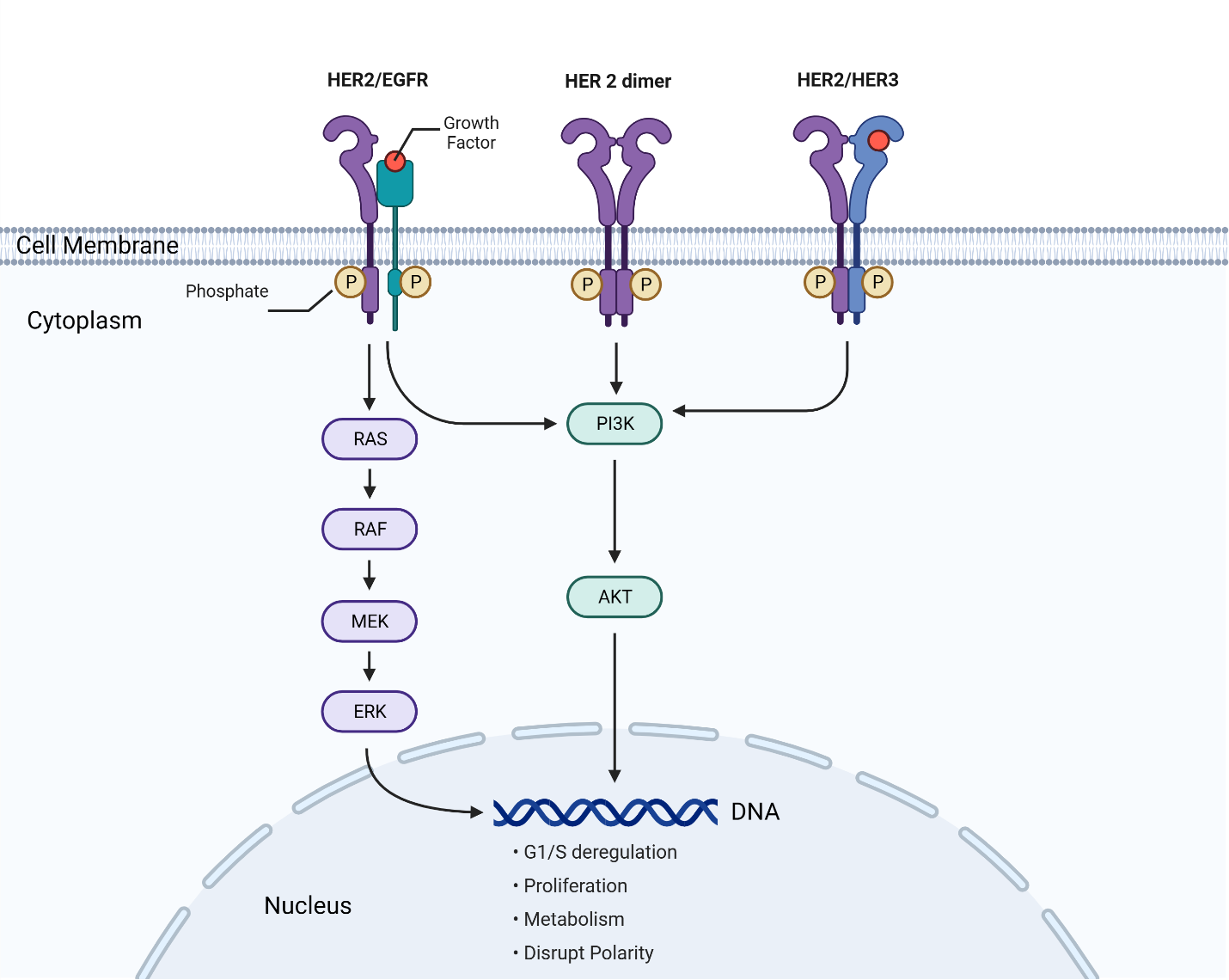
The cancer-causing effect of overexpressed HER2 receptors is due to its unique mechanism of action that involves receptor dimerization and activation of molecular signaling pathways that process growth-stimulating signals between cells (Ross et al., 2003). The HER2 protein is part of the EGFR (epidermal growth factor receptor) family and follows a similar process by binding to growth factors and initiating signal transduction. The HER2 pathway is depicted in Figure 1. The pathway involves growth factors that bind to(HER2 receptors) on cell surfaces. This triggers a cascade of signals from the receptors to proteins in the cytoplasm, eventually reaching the nucleus to activate specific genes and promote the cell cycle (National Institutes of Health (US), 2007). This involves protein phosphorylation, where one protein’s enzymatic activity triggers another’s in the signaling pathway.

In addition, HER2 signal transduction includes major pathways such as PI3K/Akt, PLC-γ, Ras-MAPK (mitogen-activated protein kinase), and JAK/STAT (Janus Kinase/Signal Transducer and Activator of Transcription). Receptors in the EGFR family, such as EGFR, HER2, HER3, and HER4, undergo dimerization to initiate phosphorylation and activate signaling pathways. The phosphorylation of specific intracellular tyrosine residues on HER2 and the subsequent signaling pathways are determined by the growth factor and dimerization partner (Ross et al., 2003).
The increase in HER2 receptors due to HER2 gene amplification increases the formation of HER2-containing heterodimers that stimulate signaling pathways regulating cell growth, leading to excessive cell growth and the development of cancer (Yarden, 2001). HER2-EGFR heterodimers initiate signaling pathways that drive cell proliferation. Increased dimers of this kind lead to deregulated G1 (growth 1 phase) and S (synthesis phase) cell cycle checkpoint control and result in uncontrolled cell proliferation. Increased HER2 homodimers initiate signaling pathways that disrupt cell polarity (Figure 1). The loss of cell polarity and cell adhesion is a common feature of many epithelial cancers (Moasser, 2007). In invasive tumors, these characteristics are common and well correlated with their metastatic ability to invade adjacent tissues (Wodarz & Näthke, 2007). HER2-HER3 heterodimers initiate the PI3K/Akt pathway that drives cell proliferation and metabolism. Increased dimers of this kind lead to uncontrolled cell proliferation (Moasser, 2007).
HER 2 Targeted Therapy
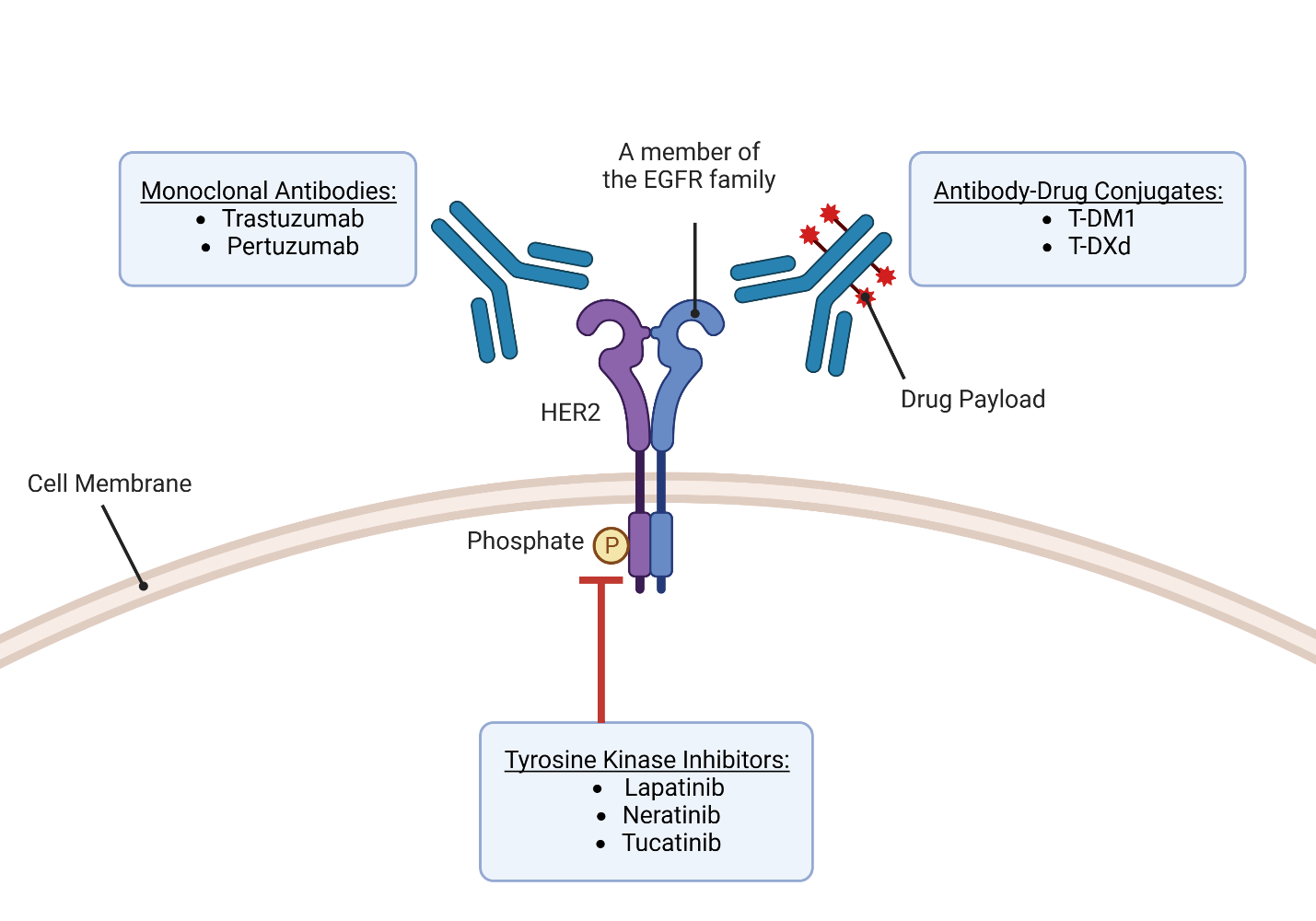
Over the past 25 years, there has been remarkable progress in the development of targeted therapies for breast cancer, which specifically inhibit pathways driving the growth and survival of breast tumors (Figure 2). This advancement has been particularly successful in HER2-positive (HER2+) breast cancer, with the introduction of drugs designed to target the overexpressed HER2 receptors. HER2-targeting therapies are now a crucial component of the first-line treatment for patients with HER2+ tumors and the combination of different HER2-targeting drugs and chemotherapy agents has demonstrated significant benefits in terms of patient outcomes. However, it is important to acknowledge that these drugs have the risk of causing serious adverse effects due to their cardiotoxicities. In general, HER2-targeted therapies can be classified into three groups based on their mechanism of action—antibodies, antibody-drug conjugates, and tyrosine kinase inhibitors (Jacobs et al., 2022).
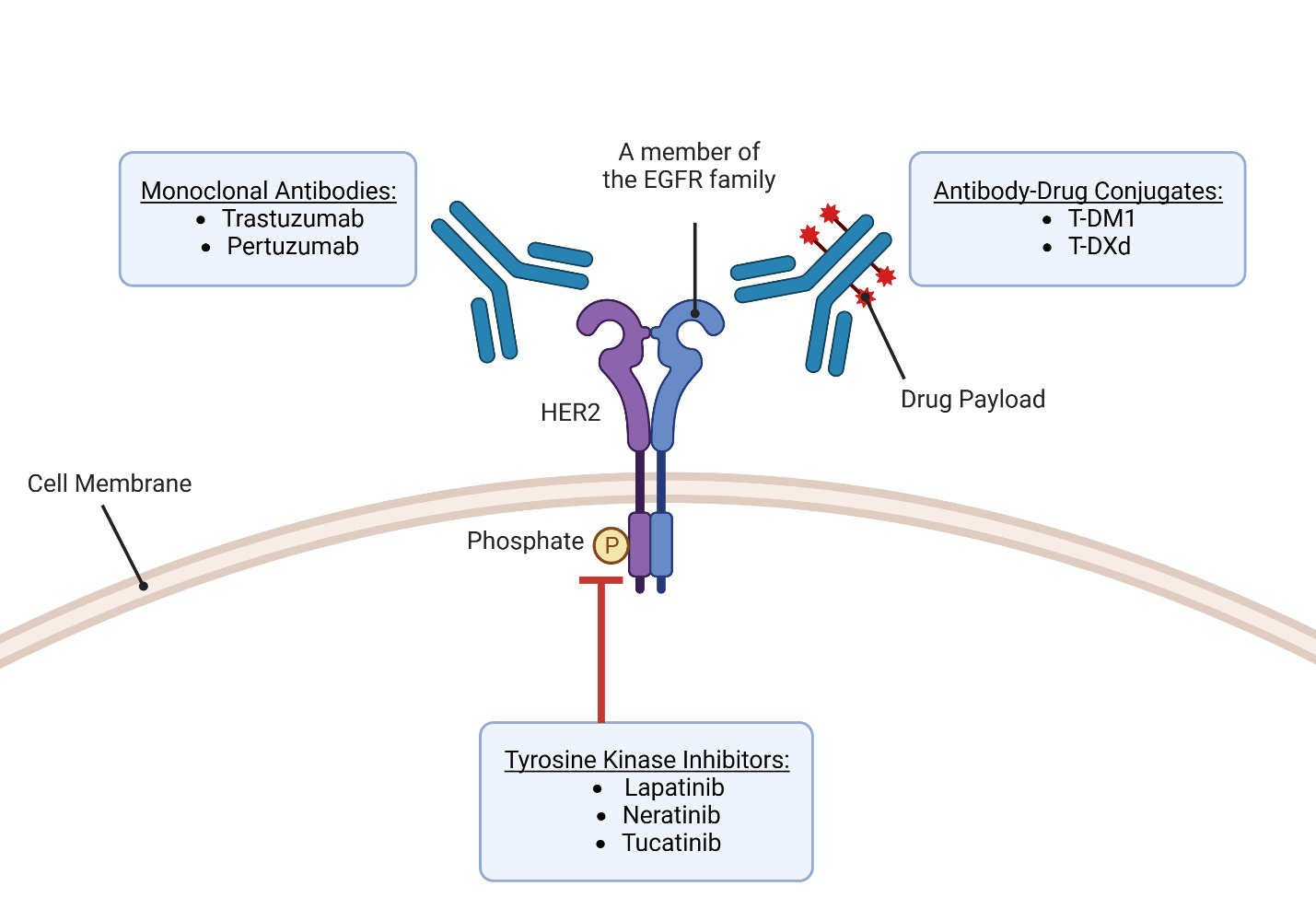
Trastuzumab
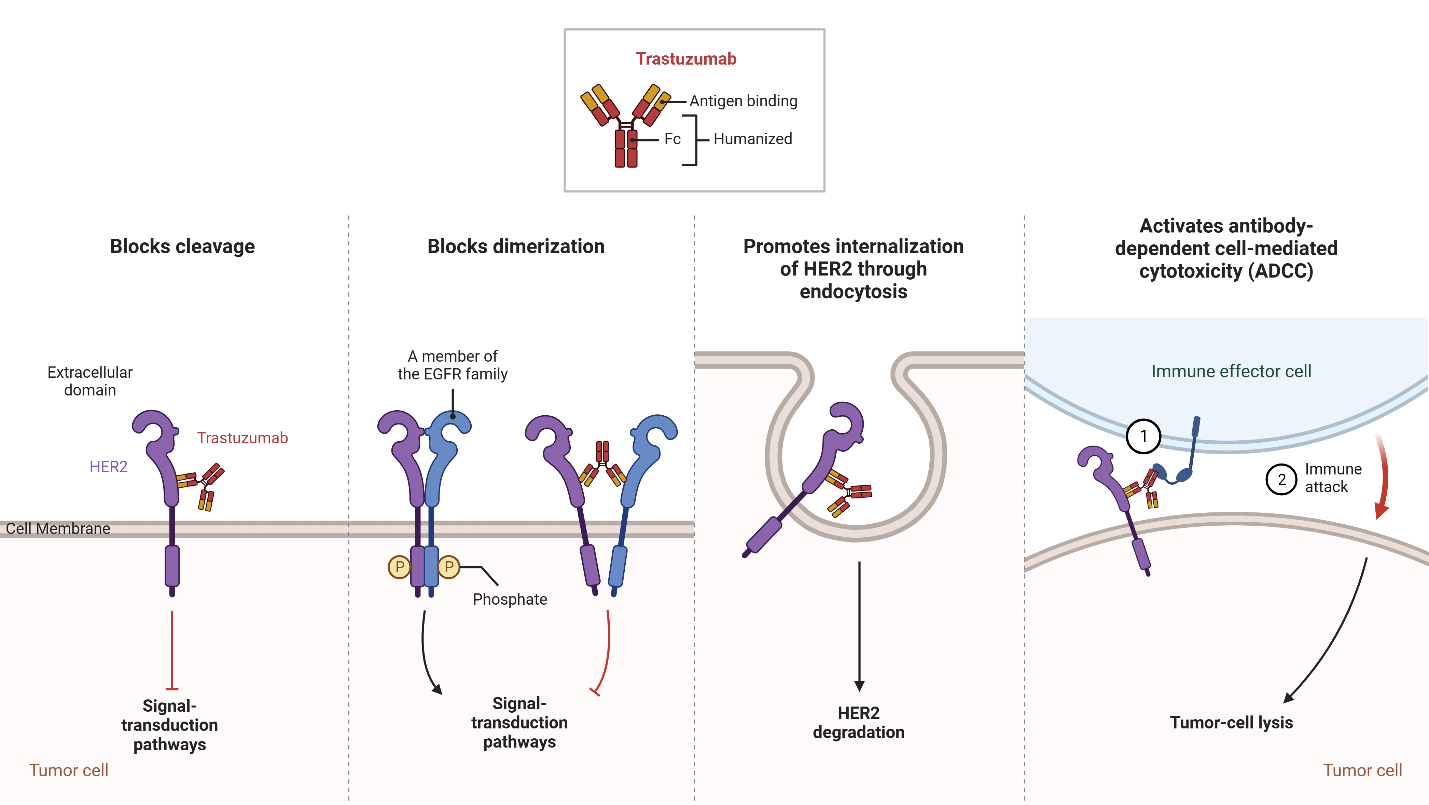
Trastuzumab, a monoclonal IgG1 class humanized murine antibody, was developed by the Genentech Corporation using recombinant technologies. Its purpose is to selectively bind to the extracellular region of HER2 in HER2+ breast cancer (Ross et al., 2003). Since its introduction in 1998, trastuzumab has emerged as a significant therapeutic choice for patients diagnosed with HER2+ breast cancer and is the first approved anti-HER2 targeted agent with promising results (F. Lei et al., 2011).
The HER2 receptor, overexpressed on the surface of HER2+ breast cancer cells, consists of different domains, including an extracellular ligand-binding domain, a transmembrane region, and an intracellular tyrosine kinase domain. Trastuzumab specifically binds to the extracellular domain of HER2 to prevent the cleavage of the extracellular domain and block receptor activation as shown in Figure 3. This inhibition interferes with the dimerization of HER2 to prevent the receptor from stimulating downstream signaling pathways that drive cell proliferation. Furthermore, trastuzumab promotes the internalization of HER2, resulting in the removal of HER2 receptors from the cell surface (Boekhout et al., 2011).
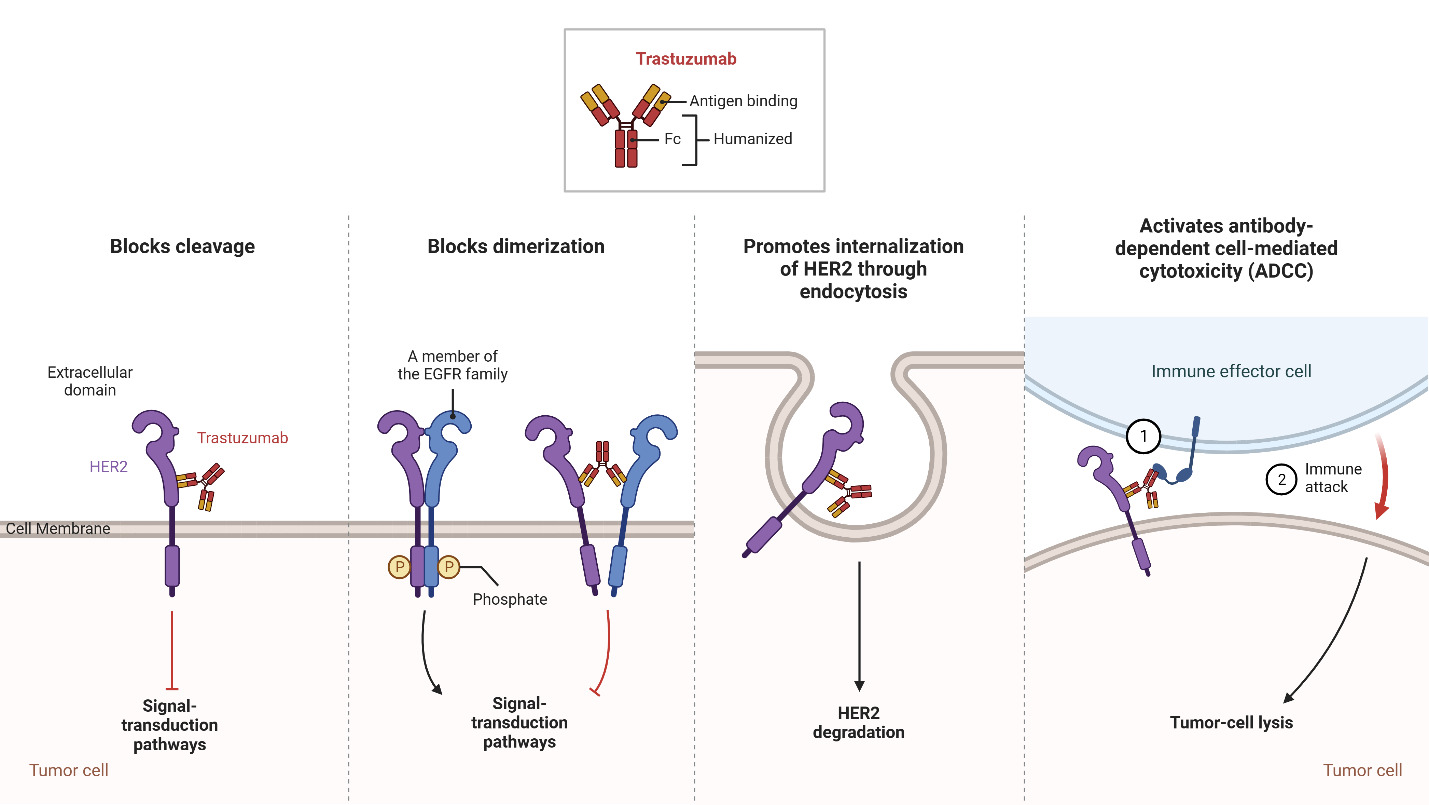
In addition to blocking receptor activation, trastuzumab also activates antibody-dependent cell-mediated cytotoxicity (ADCC), causing immune cells to recognize and destroy cancer cells with HER2 overexpression (Boekhout et al., 2011). ADCC is a mechanism that involves the targeting of cells coated with IgG antibodies, primarily the IgG1 subclass in humans. Effector cells expressing FcγRIIIa (CD16a), such as natural killer cells, monocyte/macrophages, natural killer T cells, and γδ T cells recognize the Fc region of the IgG antibodies bound to the surface of target cells (Ochoa et al., 2017). As an IgG1 class antibody that binds to HER2 receptors, Trastuzumab facilitates the engagement of immune effector cells, which leads to cell-to-cell cytolysis and the destruction of HER2+ cancer cells (Mandó et al., 2021). This immune-mediated cytotoxicity is a valuable mechanism in cancer treatment, as it enhances the body’s natural defense mechanisms against cancer and contributes to tumor regression and improved clinical outcomes. By targeting the HER2 receptor, trastuzumab provides a targeted therapeutic approach to inhibit HER2-driven tumor growth and improve clinical outcomes in patients with HER2-positive breast cancer (Zhao et al., 2021). Through the utilization of ADCC, targeted antibodies such as Trastuzumab offer a promising approach to cancer therapy, providing a specific and efficient means to target malignant cells while sparing normal cells. The combination of monoclonal antibodies with the immune system’s cytotoxic capabilities represents a significant advancement in the field of cancer treatment, offering new opportunities for personalized and targeted therapeutic interventions (Mandó et al., 2021).
Pertuzumab
Pertuzumab, developed by Genentech in 2009, is a monoclonal antibody that targets HER2, similar to trastuzumab (Luo et al., 2017). However, pertuzumab has a distinct binding mechanism compared to trastuzumab. While trastuzumab binds to domain IV of HER2, pertuzumab binds to a different region, near the center of domain II. This unique binding site allows pertuzumab to sterically block a critical binding pocket required for HER2 heterodimerization with HER3, another receptor in the HER family. By inhibiting the HER2-HER3 interaction, pertuzumab disrupts the signaling pathways involved in cancer cell growth and survival, leading to the inhibition of tumor progression (Franklin et al., 2004).
Combination therapy with pertuzumab and trastuzumab has demonstrated clinical benefits in the treatment of metastatic cancer, advanced local cancer, and early-stage cancer. The combination of these two HER2-directed monoclonal antibodies provides dual HER2 blockade by targeting different domains within the extracellular dimerization domain of HER2. This synergistic approach enhances the inhibition of HER2-driven signaling pathways, resulting in more effective suppression of tumor growth and improved patient outcomes (Ishii et al., 2019). The development and utilization of pertuzumab as part of combination therapy with trastuzumab highlight the continued progress in targeted treatments for HER2-positive breast cancer. By targeting different regions of the HER2 receptor, pertuzumab and trastuzumab provide complementary mechanisms of action and maximize the inhibition of HER2 signaling by offering a more comprehensive approach to HER2-directed therapy (Richard et al., 2016).
Antibody-drug conjugates
Antibody-drug conjugates (ADCs) represent an innovative approach in cancer therapy by combining the specificity of an antigen-targeting antibody with the potent cytotoxic effects of a payload using a molecular linker. This conjugation allows for the selective delivery of the cytotoxic agent to tumor cells expressing the target antigen, leading to the reduction of systemic toxicity (Najjar et al., 2022). In the context of HER2-positive breast cancer, ado-trastuzumab emtansine (T-DM1) and fam-trastuzumab deruxtecan-nxki (T-DXd) are the two ADCs that have been approved by the FDA for different indications (Ferraro et al., 2021).
T-DM1 was the first HER2-targeting ADC approved in 2013. Initially indicated for the treatment of metastatic patients who had previously received trastuzumab and a taxane, its label was later expanded in 2019 to include adjuvant treatment for high-risk patients with residual disease after neoadjuvant taxane and trastuzumab-based therapy (Ferraro et al., 2021). T-DM1 combines trastuzumab with the cytotoxic agent emtansine, allowing for targeted delivery of the payload to HER2-expressing tumor cells. By binding to HER2, T-DM1 initiates the internalization of the HER2-T-DM1 complex via receptor-mediated endocytosis. The non-reducible linker in T-DM1 remains stable throughout the process and ensures the integrity of T-DM1 during systemic circulation (Barok et al., 2014). Once internalized, T-DM1 undergoes proteolytic degradation in the lysosome, leading to the active release of DM1, the cytotoxic payload. The lysosomal proteases cleave the antibody part of T-DM1, liberating DM1-containing metabolites (Erickson et al., 2006). These released metabolites exert their cytotoxic effects by inhibiting microtubule assembly, thereby disrupting critical cellular processes involved in cell division and leading to cell death (Barok et al., 2014).
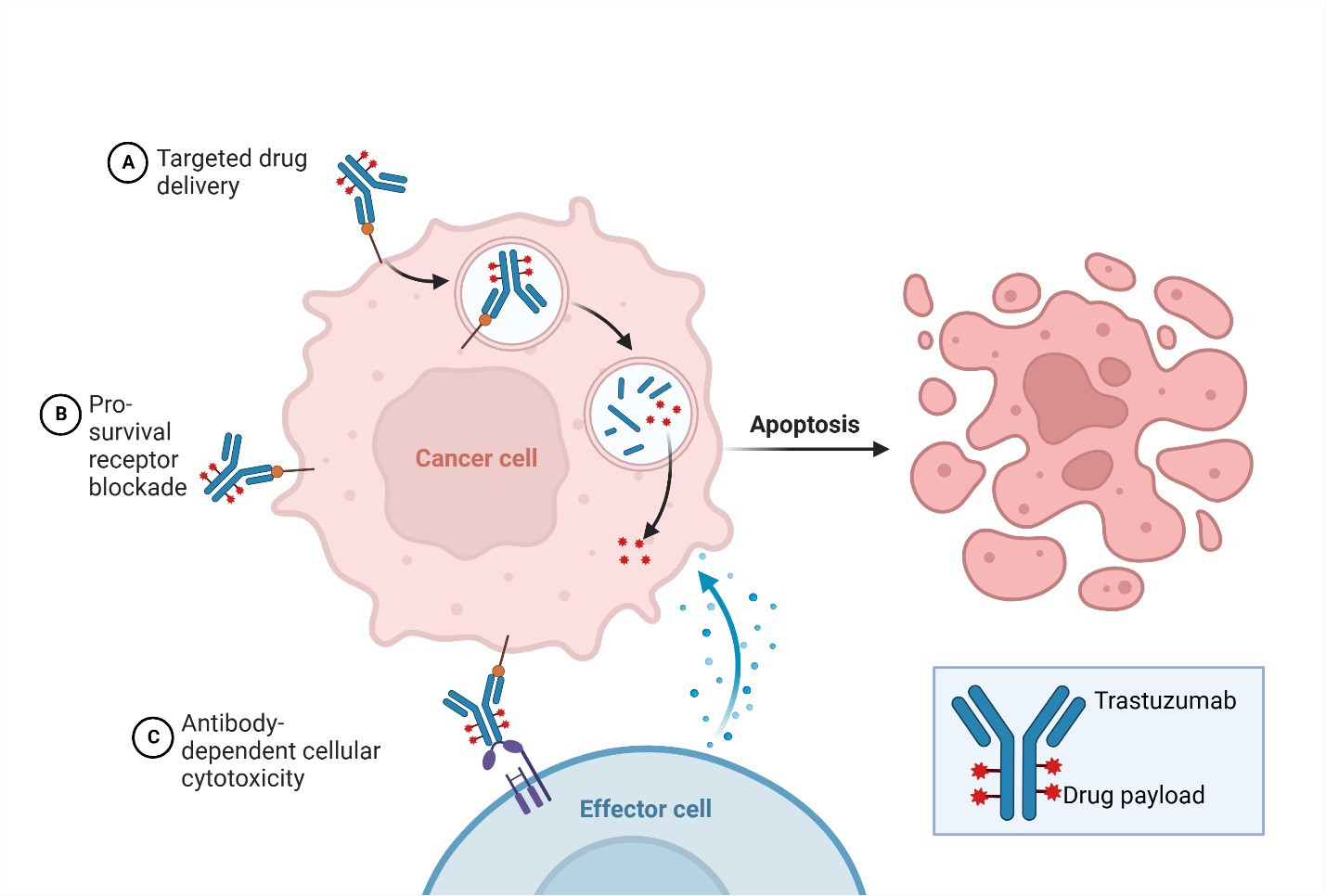
Following the approval of T-DM1, fam-trastuzumab deruxtecan-nxki (T-DXd) received FDA accelerated approval in 2019 for the treatment of patients with HER2-positive breast cancer who have received at least two prior lines of anti-HER2-based therapy in the metastatic setting (Narayan et al., 2021). T-DXd combines trastuzumab with the topoisomerase I inhibitor deruxtecan to enable targeted delivery of the cytotoxic payload to HER2-overexpressing cancer cells. Although both ADCs incorporate trastuzumab and initiate receptor-mediated endocytosis to internalize itself and release the cytotoxic payload, there are distinct pharmaceutical differences between these two ADCs that contribute to their unique mechanisms of action and therapeutic effects (Cortés et al., 2022). One notable difference lies in the drug-to-antibody ratio (DAR), which represents the number of cytotoxic payloads attached to each antibody molecule. T-DXd has a higher DAR of 8, indicating a greater number of cytotoxic payload molecules attached to each trastuzumab antibody compared to T-DM1, which has a DAR of 3.5. Furthermore, the cytotoxic payload in T-DXd is derived from exatecan, a topoisomerase I inhibitor (Ferraro et al., 2021). In contrast, T-DM1 utilizes a microtubule inhibitor as its cytotoxic payload (Barok et al., 2014). Topoisomerase I inhibitors interfere with the activity of the topoisomerase I enzyme, which is involved in DNA replication and transcription. These inhibitors bind to the enzyme’s active site and prevent the resealing of DNA strands after they are cleaved (Rothenberg, 1997). This leads to the formation of covalent complexes between the enzyme and DNA that cause DNA damage and disrupt essential cellular processes. This property allows T-DXd to inhibit cell proliferation in HER2+ cancer cells and induce cell death (Pommier, 2009).
The difference in the type of cytotoxic agent used provides an alternative strategy for targeting HER2+ breast cancer cells by disrupting different critical cellular processes necessary for their survival. The approvals of T-DM1 and T-DXd highlight the clinical success of ADCs in treating HER2-positive breast cancer, offering new options for patients who have progressed on previous therapies. By harnessing the specific targeting ability of antibodies, leveraging distinct cytotoxic payloads, and optimizing the drug-to-antibody ratio, ADCs like T-DXd and T-DM1 offer promising treatment options that specifically target HER2+ tumor cells, potentially improving patient outcomes in this subset of breast cancer.
Tyrosine Kinase Inhibitors
Kinases are crucial enzymes that play a significant role in cell signaling and various cellular processes by transferring phosphate from ATP to specific amino acid residues. In numerous cancers, there is evidence of kinases, particularly receptor tyrosine kinases, being overexpressed (Chaar et al., 2018). In HER2+ breast cancer, the overexpression of the tyrosine kinase receptor HER2 makes it an attractive target for therapeutic intervention through the use of tyrosine kinase inhibitors (TKIs) (Du et al., 2021). HER2 possesses the small side chain amino acid threonine at its gatekeeper site (Azam et al., 2008). This characteristic creates a hydrophobic pocket near the ATP-binding site that can be targeted by TKIs. However, this structural feature also renders HER2 vulnerable to drug resistance because a gatekeeper mutation substituting the small threonine with an amino acid featuring a larger side chain can confer resistance to TKIs (Cohen et al., 2021).
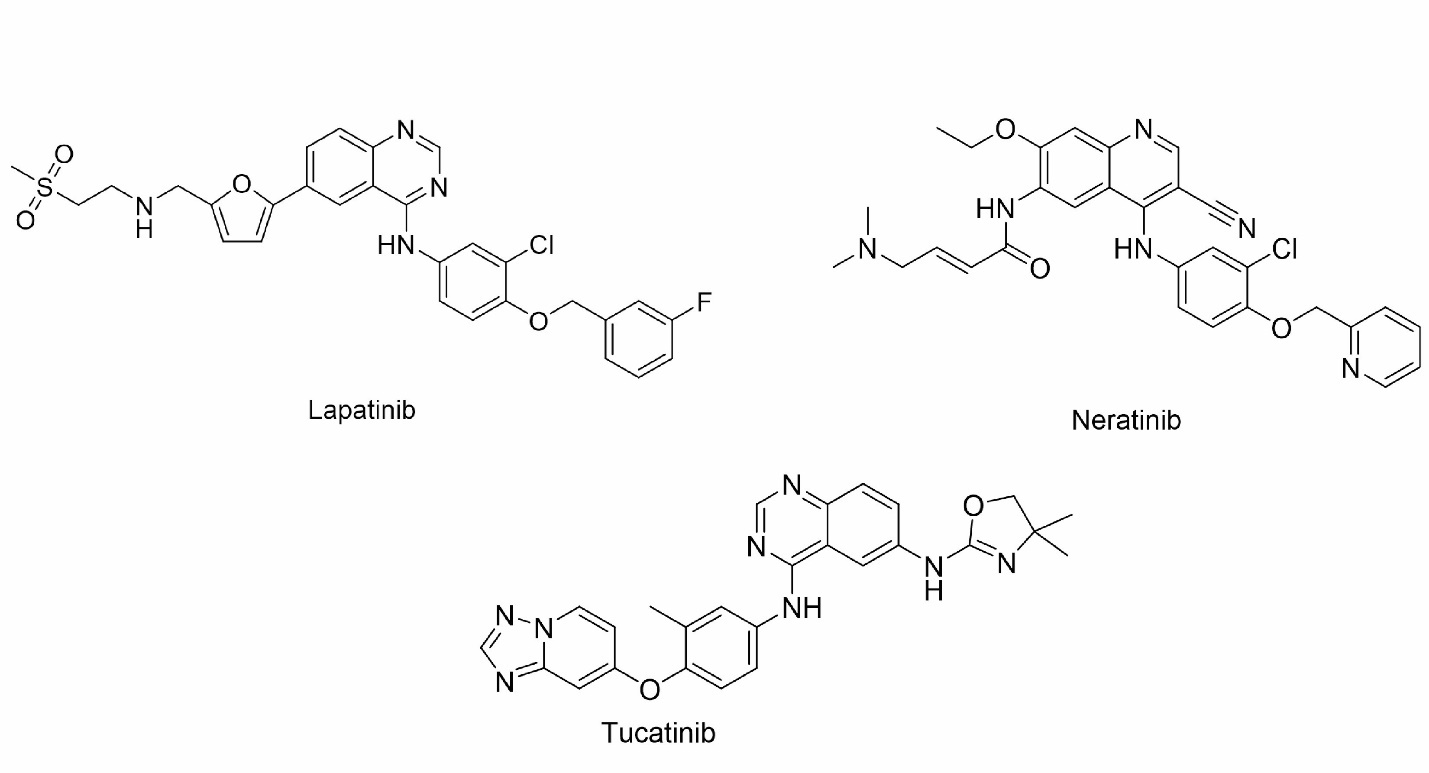
TKIs are oral non-peptide anilinoquinazoline compounds that are designed to compete with ATP for binding to the ATP-binding domain of protein kinases. By binding to the kinase domain, TKIs prevent the phosphorylation and subsequent activation of downstream signaling pathways, leading to the inhibition of cell proliferation and induction of apoptosis (Segovia-Mendoza et al., 2015). One of the advantages of TKIs is their ability to target multiple members of the human epidermal growth factor receptor (EGFR) family involved in important signaling pathways. These pathways include PI3K/AKT, MAPK, PLC γ, ERK1/2, and JAK/STAT, which are critical for cell survival, proliferation, and differentiation. Through their broad inhibitory effects on these pathways, TKIs can impact the expression of various transcription factors such as MDM2, mTOR, and p27 to further regulate cellular processes involved in cancer development and progression. Lapatinib in 2007, neratinib in 2017, and tucatinib in 2020 are the three TKIs approved by the FDA to treat HER2+ breast cancer (X. Yang et al., 2020). The molecular structures of these three TKIs are shown in Figure 5. The development of small molecule TKIs represents a significant advancement in targeted cancer therapy, providing a more specific and tailored approach to inhibit aberrant kinase activity. By interfering with critical signaling pathways and transcription factors, TKIs hold promise as effective therapeutic agents for various cancers, offering the potential to inhibit tumor growth, induce apoptosis, and improve patient outcomes.

Chemotherapy
Principles of Chemotherapy
Cancer treatment is a complex process based on three primary principles. The first principle is the fraction-kill hypothesis, which states that a uniform drug dose kills a constant fraction of tumor cells regardless of the tumor burden. The second principle is that tumor cells respond linearly to the dose of the administered drug. The third principle is the Goldie-Coldman hypothesis, suggesting that cancer cells develop spontaneous mutations leading to drug resistance. Due to these principles, combination or multitargeted therapy is more effective than using a single agent in cancer treatments. Using chemotherapy agents with different mechanisms of action and nonoverlapping toxicities can lower drug resistance and target multiple aspects of the cell cycle by affecting both resting and dividing cells (Amjad, 2023). Chemotherapy serves various goals in cancer treatment. Curative chemotherapy aims to eliminate all cancer cells from the body (X. Yang et al., 2020). Adjuvant chemotherapy is commonly used for breast cancer and targets any remaining cancer cells after the initial treatment to prevent recurrences (Amjad, 2023). Neoadjuvant chemotherapy is used when a tumor is too large for direct surgery, shrinking it for complete surgical removal or less invasive procedures. Palliative chemotherapy is given when complete removal of tumor cells is not possible, aiming to relieve symptoms, slow disease progression, and avoid complications (X. Yang et al., 2020).
Alkylating Agents
Breast cancer is a prevalent form of cancer worldwide, and alkylating chemotherapy agents such as cyclophosphamide are commonly used to treat it (Hung et al., 2017). Alkylating agents work by inhibiting the transcription of DNA into RNA, resulting in the cessation of protein synthesis. Through the alkylation process, alkyl groups replace hydrogen atoms in DNA, forming cross-links within the DNA chain that have cytotoxic, mutagenic, and carcinogenic effects. Alkylating agents affect all cells, but they primarily target rapidly dividing cells that lack sufficient time for DNA repair, such as cancer cells. This alkylation process causes misreading of the DNA code, inhibits DNA, RNA, and protein synthesis, and triggers apoptosis in rapidly proliferating tumor cells. The cytotoxic effects of alkylating agents lead to the destruction of rapidly dividing cancer cells, resulting in reduced tumor size and burden. However, other rapidly dividing cells, such as hematopoietic cells, reproductive cells, and endothelial cells, are also significantly affected by this type of chemotherapy. Common side effects of alkylating agents include anemia, pancytopenia, amenorrhea, impaired spermatogenesis, intestinal mucosal damage, alopecia, and an increased risk of malignancy. While these side effects can be significant, their potential benefits in treating cancer often outweigh the risks (National Institute of Diabetes and Digestive and Kidney Diseases, 2015).
Antimetabolites
Although less popular, antimetabolite chemotherapy agents such as methotrexate are still used to treat breast cancer (V. Yang et al., 2020). Antimetabolites are a class of chemotherapy agents that interfere with the synthesis of DNA nucleotides. They can be either of purine and pyrimidine bases or of folate cofactors, which are involved in several steps of purine and pyrimidine biosynthesis. By inducing depletion in nucleotides, they inhibit DNA replication in the S phase of the cell cycle. Some antimetabolites can also be fraudulently inserted into nucleic acids to induce structural abnormalities leading to cell death, such as DNA breaks (Lansiaux, 2011). However, antimetabolites can cause several side effects, with myelosuppression being the most common. Myelosuppression is the suppression of stem cells in the bone marrow, which leads to a decrease in the number of red and white blood cells and platelets, causing anemia, leukopenia, and thrombocytopenia (Amjad, 2023).
Antimicrotubule Agents
Antimicrotubule chemotherapy agents such as anthracyclines and taxanes are commonly used to treat breast cancer. However, like many other chemotherapy agents, common side effects of anthracyclines and taxanes include anemia, leukopenia, and thrombocytopenia caused by myelosuppression. Antimicrotubule agents disrupt the M phase of the cell cycle and drive the cell to undergo apoptosis. As an antimicrotubule agent, taxanes such as paclitaxel, docetaxel, and cabazitaxel, disrupt the balance of microtubule polymerization and depolymerization, resulting in abnormal cellular function. Taxanes inhibit microtubule assembly to disrupt cell replication and cause apoptosis in the M phase of the cell cycle (Amjad, 2023). Being another antimicrotubule agent, anthracyclines inhibit topoisomerase II (topII)—an enzyme involved in uncoiling DNA. Through this inhibition, anthracyclines cause DNA strand breaks and cell apoptosis (Saleh et al., 2020). In addition to taxanes and anthracyclines, other antimicrotubule agents such as vinca alkaloids (vinblastine, vincristine, and vinorelbine) bind to tubulin and inhibit microtubule formation during the metaphase of mitosis (Amjad, 2023).
Prospects for Breast Cancer Treatment—Artificial Intelligence
AI offers promising opportunities to enhance breast cancer detection, diagnosis, and treatment by leveraging its ability to analyze vast amounts of data and provide valuable insights to healthcare professionals and researchers (Tran et al., 2019). AI has the potential to significantly improve the accuracy and efficiency of breast cancer detection and diagnosis. Computer-aided detection (CAD) systems, powered by AI algorithms, can analyze mammograms and other imaging modalities to identify suspicious areas that may require further investigation. By flagging potential abnormalities, AI can assist radiologists in reducing false negatives or positives, leading to more accurate and timely diagnoses (Zheng et al., 2023). Moreover, AI algorithms can process large amounts of complex medical data, including patient records, imaging data, genetic information, and pathology slides. By extracting patterns and insights from this data, AI can aid researchers and clinicians in identifying potential risk factors, diagnosing breast cancer at an early stage, and predicting patient outcomes (Dileep & Gyani, 2022). In the field of drug development for breast cancer, AI is also playing a significant role. Machine learning algorithms can analyze extensive biomedical literature, genomic data, and drug databases to identify potential drug targets and predict the efficacy of specific compounds. This technology can facilitate virtual screening, optimize lead compounds, and design more targeted and efficient therapies, potentially accelerating the drug development process (Liang et al., 2020).
Conclusion
HER2 receptor overexpression caused by HER2 gene amplification in chromosome 17 is a common cause of breast cancer. When HER2 receptors bind to growth factors, they dimerize with other members of the EGFR family and activate signaling pathways that drive cell proliferation. Treatment options for HER2+ breast cancer include generic chemotherapy drugs like alkylating agents, antimetabolites, and antimicrotubule agents. Targeted therapies for HER2+ breast cancer include monoclonal antibodies that selectively bind to HER2 receptors to block receptor activation and activate antibody-dependent cell-mediated cytotoxicity (trastuzumab, pertuzumab), antibody-drug conjugates that have cytotoxic drug payloads attached to monoclonal antibodies (T-DM1, T-DXd), and tyrosine kinase inhibitors that disrupt the ATP binding kinase domains of the HER2 receptor (lapatinib, neratinib, tucatinib). For maximum efficacy, combination therapy involving multiple drugs with different mechanisms of action is commonly used. For the future of breast cancer, artificial intelligence holds promise in aiding breast cancer detection, diagnosis, and drug development due to its data analysis capabilities.
Author Note
I have no conflict of interest to disclose.